Moleküler Dinamik Simülasyonları Bileşenleri¶
EpiSTEM Türkiye'deki yazıyı takiben biraz daha detaya girelim nedir bu simülasyonlar. Bileşenleri nelerdir?
Yazımda da değindiğim gibi Newton'un kanunlarından yola çıkılmıştır moleküler dinamik simülasyonları için. Bir cismin hareketi ona uygulanan kuvvet ile orantılıdır. Elinizle bir objeyi ne kadar uzağa fırlatabileceğiniz ona uyguladığınız kuvvet ile belirlenir. Moleküler dinamik simülasyonlarında da bir molekülün hareketi de ona uygulanan kuvvete bağlıdır. Bu kuvveti tanımlarken molekülün sahip olduğu geometrik yapıyı ve çevresini saran diğer molekülleri baz alıyoruz. İşte bu aşamada kuvvet alanı dediğimiz bir grup özellikler devreye giriyor.
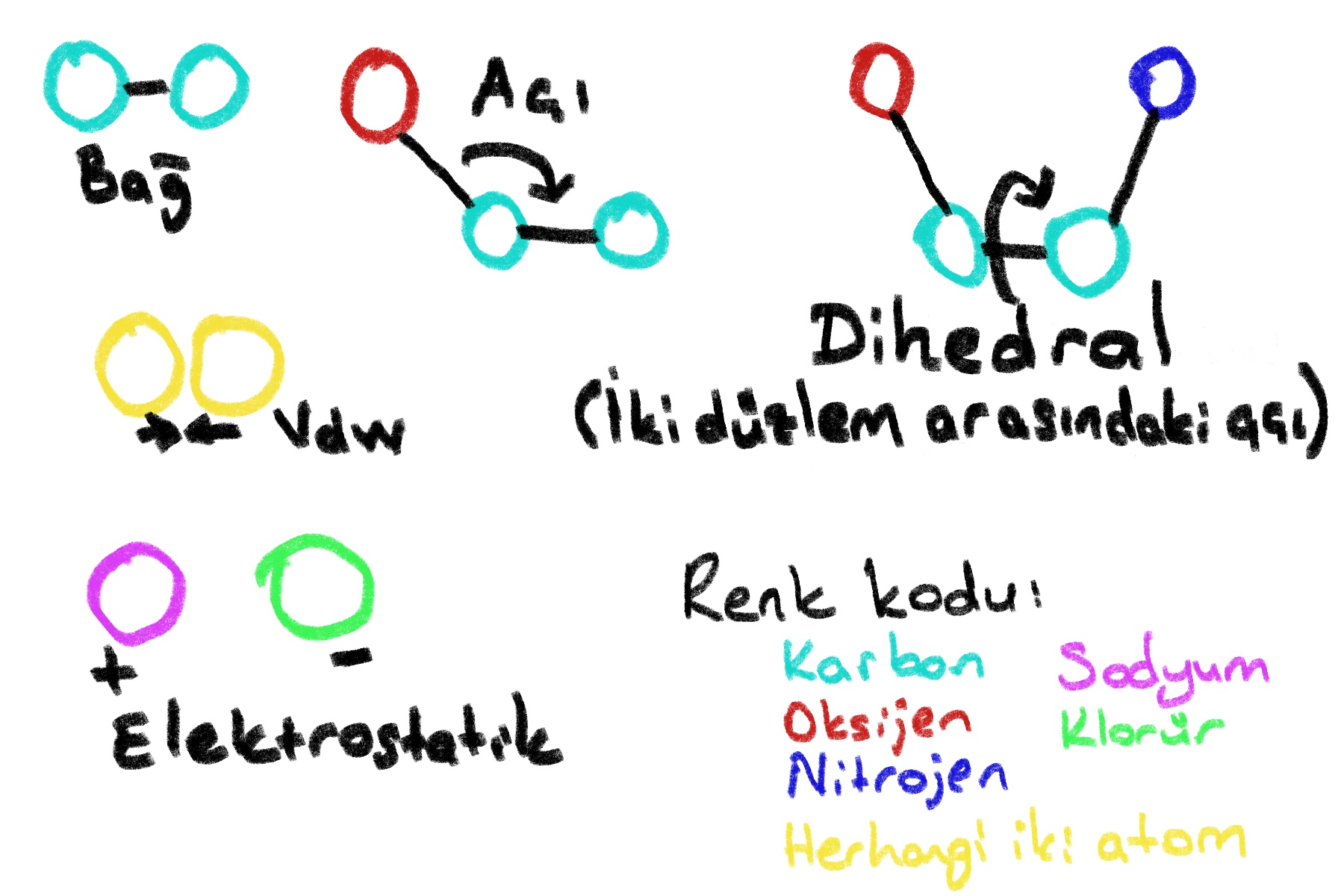
Nasıl insanların kol uzunlukları farklı ise, atomları birbirine bağlayan bağlarda farklı uzunluklara sahiptirler. Kimimizin kolu çabuk kırılırken, kimimiz sağlam. Bu bağların dayanıklılığı da aynı böyle değişkendir. Meraklısına aşağıdaki grafik bir kuvvet alanındaki tanımlı özellikleri özetliyor. Grafiğin uyarlandığı kaynağı okumanızı tavsiye ederim (Maalesef İngilizce).
Kuvvet alanı moleküler dinamik simülasyonlarının en temel ve en önemli bileşenleridir. Kuvvet alanına ek olarak bilmemiz gereken bir diğer bileşen ise koordinat. Yani atomların adresleri. Hepimizin bir ev ve iş adresi var. Koordinatlarıda atomların yerini belirleyen adresler olarak düşünebilirsiniz. Bu koordinatlar elbette 3 boyutlu. Basit moleküllerin koordinatlarını tahmin yolu ile çizmek mümkün olabilir. Fakat 5 tane yapı taşından (amino asitten) oluşan proteinin bile alabileceği çok sayıda şekil var. O nedenle deneysel yöntemlere başvuruyoruz bu koordinatları elde ederken.
İhtiyacımız olan üçüncü bileşen ise simülasyonu çalıştıracağımız program. Benim deneyimlerime göre GROMACS simülasyonlara yeni başlayanlar için en güvenilir program. Komut isteminden kullanmak birçok kişiyi ürkütse de yeni başlayan biri olarak yapacağınız hataların geri bildirimini almak çok önemli ve GROMACS bu konuda en iyilerden biri diyebilirim. Kullanabileceğiniz diğer bir alternatif NAMD, ki bunu VMD programından bir arayüz ile kullanabilirsiniz. Görsel açıdan öğrenmesi çok daha kolay bir program. Bir diğeri ise bir çok özelliği barındıran bir nevi kodlama dili diyebileceğimiz CHARMM; simülasyon yapabilirsiniz ama öğrenmesi kolay olmayan bir programdır kendisi (hata mesajlarının yoksunluğu da cabası). Ama GROMACS'a kıyasla sistemler üzerinde değişiklik yapması daha kolay bir program. Amber ismi verilen programına dair bir fikrim yok, çünkü hiç kullanmadım, ama ismi burada dursun ki sizin de haberiniz olsun. Lakin Amber'in aynı isimli kuvvet alanını kullandım ki bu rehberde onun üzerine. Alternatif kuvvet alanı arıyorsanız, CHARMM isimli (yukarıda bahsettiğim programı geliştirenler tarafından hazırlanan) kuvvet alanına bakabilirsiniz.
Programı belirledik, GROMACS. Kuvvet alanımız ise, Amber. Kaldı geriye koordinatlar... Bunun için PDB veri bankasına gidip PDB formatında bir protein yapısı indirdim: İnsülin. Linkten daha fazla bilgi edinebilirsiniz. Bu proteini seçmemin sebebi, muhtemelen bir çoğunuzun ismini duymuş olması. Şeker hastası var mı aramızda? Kendinize enjekte etmekte olduğunuz insülin ve insülin benzeri proteinler sayesinde hayat kaliteniz biraz daha artıyor, artık kendisi ile moleküler düzeyde de tanışmış oldunuz.
import nglview #NGLview paketini ortama yükle (https://github.com/arose/nglview)
view = nglview.show_pdbid("2ins") #"2ins" kodlu yapıyı PDB veri bankasından indir ve NGLview uygulamasına gönder
view #görüntüle
%%html --isolated
<iframe src="2INS.html" allowTransparency="true" scrolling="no" width=100% height=400px> </iframe>'
Yukarıda görmüş olduğunuz yapı insülinin aktif formu. Proteinlerin aktif formları birbirinin aynı veya farklı bir çok zincirden oluşabilir. Hız kazanmak açısından bu rehberde biz sadece aşağıda göreceğiniz tek bir zincir ile devam edeceğiz. Bu arada üstteki resim hareketli. Başka bir html sayfayı yükleyerek size bu interaktif görseli sundum, ama eğer bu rehberi indirip kendiniz çalıştıracaksanız html ile başlayan hücreleri silip python komutundan devam edebilirsiniz (Aaa söylemedim mi bu rehber Python programlama dili ile çalışıyor).
Bu rehberi hazırlarken kullandığım programlar ile ilgili bilgileri yazının en sonunda biraz anlattım. Benim hazırladığım dosyalara ve bu programlara nereden ulaşabileceğiniz de orada.
1. Simülasyon ortamının hazırlanması¶
a. Protein yapısının temizlenmesi¶
Her ne kadar sizi burada angarya dediğim bu kısım ile uğraştırmayacak olsam da bilmeniz gerek. Protein veri bankasından indirip de öyle hemen simülasyon başlatabileceğiniz yapı sayısı azdır. Bunun en birincil nedeni deneysel yöntemlerin her atoma konum atamakta eksik kalmasından kaynaklı. Günümüzde detayına inmediğim bu yöntemler (X-ray, NMR... vs.) daha yüksek kalitede protein resimleri çizebiliyor olsa da proteinin her bölgesi görüntülenemeyebiliyor. İkincil neden protein ile beraber başka moleküllerin de resminin çıkması ve bizim bunları simülasyonda istemiyor olmamız. Bu moleküllerden bazıları deneyin başarılı olması için gerekli (mesela deterjanlar), bazıları da proteinlere bağlanan ilaç gibi diğer bileşenlerdir. Genelde deterjan gibi bileşenleri simülasyonda istemiyoruz, çünkü proteinin doğal ortamında aslında onlar yok. İlaç benzeri molekülleri ise kuvvet alanında tanımları olmadığı için çıkarmanız gerekebilir (veya tanımlayarak kuvvet alanına eklememiz gerekir). Diğer nedenler ki bunlar aslında ikincil nedenden daha fazla sıklıkta görülüyor, proteinin yapı taşlarının (amino asitlerin) numaralandırılmasının farklı olması, bazı bağların eksik olması, amino asitlerin eksik olması ama eksik olarak rapor edilmemiş olması, vs. vs. Hayatınızı kolaylaştırmanın tek yolu bir protein ailesi ile çalışmaya başlamadan önce numaralandırma ve doğal amino asit dizisi kontrollerini yapıp aklı selim bir rota belirlemeniz. Ne çok laf ettin diyeceksiniz, inanın bu yapıları hazırlamak simülasyonların çalışmasından daha uzun sürüyor. Bu aşamada bir hata yapmış iseniz... hoooop... bütün veriler çöpe. Ben size aşağıda, buyurun, hazır temizlenmişini sunuyorum.
import nglview
view = nglview.show_file("2ins_zincirAB.pdb")
view #görüntüle
%%html
<iframe src="2INS_zincir.html" allowTransparency="true" scrolling="no" width=100% height=400px> </iframe>
b. Protein yapısının kuvvet alanı ile hazırlanması¶
Fark ettiniz mi hala iki zincir var insülinde. Aslında tek bir peptit zincirinden oluşur insülin, yani A ve B zincirleri tek bir gen tarafından üretilmiştir, ancak insülün aktif halini almadan önce bazı değişimlerden geçer ve sonucunda sanki birbirinden bağımsızmış gibi iki zincir oluşur. Biz şimdi bu iki zinciri kuvvet alanındaki tanımları ile "topoloji" denilen bir dosyaya yeniden yazacağız. PDB gibi dosyalar proteindeki atomların uzaydaki yerini belirlerken, topoloji bu atomların birbirleri ve çevresi ile ilişkisi hakkında nicel bilgiler içerir. Karbon atomu, oksijenden 0.15 nanometre uzakta (evet geçiniz kilometreyi santimetreyi biz nano boyuttayız artık, yani $10^{-9}$m) ve oksijen dışında 1'i hidrojen olmak üzere 3 atoma daha bağlı. Karbon-hidrojen-oksijen açısı 120 derece. ... Bu şekilde bir sürü bilgi var, bunların el ile hazırlanması mümkün olsa da, aklımızı peynir ekmek ile yemedik bu devirde. Sağ olsunlar simülasyon programlarını yazanlar bize bu dosyaları da hazırlayacak bir bileşen eklemişler. Aşağıdaki komut ile bu dosyayı çok kolay hazırlayabiliriz. Bu komutları bir komut istemi (terminalde) doğrudan çalıştırıyorum. Farklı terminaller var, benim burada kullandığım terminam bash.
%%bash
export GMX_MAXBACKUP=-1
pdb2gmx -f 2ins_zincirAB.pdb -o kuvvetli_protein.pdb -p topoloji.top -ff amber99sb-ildn -water tip3p -merge all -quiet
Bu komutta -f ile girdiyi, -o ile çıktıyı tanımladık (bu atomları kuvvet alanına göre isimlendirilmiş bir dosya). -p topoloji dosyasını tanımladı, -ff kuvvet alanını, -water su modelini seçti (anlaşılması en zor molekül sudur, birden fazla modeli vardır. Onur'un yazısından daha fazla bilgi edinebilirisniz su ile ilgili. ). -merge all ise iki zincirin topoloji bilgisini tek bir dosyaya (topoloji.top) yazdı. Üstteki seyir defterini (log) incelerseniz, insülinin toplam yükünü (charge) öğrenebilirsiniz.
c. Simülasyon kutusu ve çözelti¶
Simülasyonları bir deney tüpü olarak düşünebilirsiniz. Sadece bir sürü protein ile aynı anda deney yapmak yerine biz her bir proteini tek tek tüplere yerleştiriyoruz. Bu tüplerin sınırını biz çiziyoruz. Gönül isterdi ki kocaman hazırlayalım bu simülasyonları ama çok pahalı bir hesaplama olduğu için mümkün olduğunca ufak hazırlıyoruz. Aşağıdaki komutlar önce kutunun sınırlarını çizip, sonra da o kutuyu su ile dolduracak. Bir önceki aşamada seçtiğimiz su modeli de burada devreye giriyor. Kutunun büyüklüğü proteinin etrafında yaklaşık 1 nanometre boşluk kalacak şekilde 5.4 nanometre olarak seçildi. Su ekledikten sonra ise sistemin toplam yükünü sıfırlıyor (1) ve belirli miktarda bir çözelti hazırlıyoruz (2).
1) Atomlar arasındaki etkileşimi hesaplamada kullanılan yöntem sistemin toplam yükünün sıfır olmasını gerektiriyor. Bu yük dengeleme işi sisteme iyonlar eklenerek yapılıyor. İnsülinin yükünü öğrendi iseniz sisteme artı yüklü sodyum ($Na^{+1}$) mu yoksa eksi yüklü klorür ($Cl^{-1}$) mü eklememiz gerekiyor tahmin edebilirsiniz, çünkü kutuya eklenen suyun toplam yükü sıfır. Yani sistem insülinin yükünü taşıyor.
2) İnsülinin içinde bulunduğu doğal ortamı sistemi mümkün olduğunca basit tutarak hazırlıyoruz. Bunun en temel yolu iste sisteme daha fazla iyon yüklemek. Yani sadece net yükü sıfırlamakla kalmıyoruz, üzerine daha fazla sayıda sodyum ve klorür ekliyoruz.
-p topoloji.top bu dosyanın devamlı güncellenmesini sağlıyor, böylece sistemde ekleme/çıkarmalar sonucunda ne kaldı biliyoruz. Bu işlem önemli çünkü topoloji dosyasının PDB dosyası ile uyuşması gerek.
%%bash
export GMX_MAXBACKUP=-1
editconf -f kuvvetli_protein.pdb -box 5.4 5.4 5.4 -bt cubic -o kutuda_protein.pdb -quiet
genbox -cp kutuda_protein.pdb -p topoloji.top -cs -o sulu_protein.pdb -quiet
İyonları eklemeden önce sisteme bakalım mı? Üstteki seyir defterinde kaç tane su molekülü eklenmiş bulabilirsiniz. Aşağıda da sistemin görsel halini inceleyebilirsiniz...
import nglview #bu komut üstte çalıştığı için tekrar çalıştırmaya gerek yok ama üstteki veri kayboldu ise tekrar yükledim.
view = nglview.show_file("sulu_protein.pdb")
view.add_ball_and_stick("sol",opacity="0.5")
view #görüntüle
%%html --isolated
<iframe src="Simulasyon_sistemi_yuklu.html" allowTransparency="true" scrolling="no" width=100% height=600px> </iframe>
İyonları eklerken GROMACS'ın bir gerekliliği var, o da çalıştırma dosyası yaratmak. Bunun detaylarına girmiyorum, her simülasyon programının farklı bir yolu var bunun için. Ama aynı işlevi görüyorlar: rastgele su moleküllerini iyonlar ile değiştirmek. Yani bu aşamada sistemden su çıkararak iyonlara yer açıyoruz. Eklediğim iyon çözeltisinin miktarını aşağıdaki komut isteminden tahmin edebilirsiniz (birim mol/litre).
%%bash
export GMX_MAXBACKUP=-1
touch 4ion.mdp
grompp -f 4ion.mdp -c sulu_protein.pdb -p topoloji.top -o 4ion.tpr -quiet
genion -s 4ion.tpr -conc 0.15 -neutral -o cok_iyonize_protein.pdb -p topoloji.top -quiet << EOF
13
EOF
import nglview
view = nglview.show_file("cok_iyonize_protein.pdb")
view.add_surface("sol and not within 3 of protein",color="cyan",opacity=0.1)
view.add_ball_and_stick("NA",radiusSize=1) #mor renkli
view.add_ball_and_stick("CL",radiusSize=1) # yeşil renkli.
view #görüntüle
%%html --isolated
<iframe src="Simulasyon_sistemi.html" allowTransparency="true" scrolling="no" width=100% height=600px> </iframe>
2.Simülasyon¶
Bu kısım için maalesef size "bende hazır yapılmışı var" diyeceğim. Ama size tabii ki bu simülasyonu nasıl elde ettiğimi anlatacağım. Ben bu rehberi genişletene kadar sizi İngilizce de olsa başka bir kaynağa yönlendirmek istiyorum. Justin Lemkul bu rehberleri GROMACS için hazırlamış ve düzenli olarak güncelliyor. Farklı bir sürü örnek bulabilirsiniz simülasyonlar ile neler yapılabileceğine dair. Dönelim bizim insüline...
Sağlıklı bir veri elde etmemiz için öncelikle sistemi biraz rahatlatmamız lazım. Biz proteini hazırlarken, suyu ve iyonları eklerken hiç sormadık acaba keyfiniz yerinde mi, komşu atomlarınız sizi çok rahatsız ediyor mu diye. Bu aşamada sistemdeki her bir bileşenin hem kendi içindeki hem de çevresindeki kavgalarını çözeceğiz. Bir diğer deyişle, enerjisini azaltacağız. Rahatlatma birbirini takip etmesi gereken iki ayrı basamak ile sağlanıyor.
- Enerji İndirgenmesi: Protein yapıları protein veri bankasına yüklenirken bizim kullandığımız kuvvet alanları ile hazırlanmadıkları için sahip oldukları enerji yüksek olabiliyor. Bu enerji, yazının başındaki grafikteki terimler ile hesaplanıyor. Sistemin enerjisi yüksek olduğunda hareketi de aşırı olacaktır. Biz zaten proteini hareket ettirmek istiyorduk diyeceksiniz ama bu aşırı hareket fiziki olmayan bir durum. Mesela bizim tanımladığımız hidrojen atomları (deneysel yöntemler hidrojen atomlarını göremezler) diğer atomlara gereğinden fazla yakın yerleştirilmiş olabilir. Bu nedenle bu aşamada her molekül bulunduğu yerde titreştiriliyorlar (aynen telefonun titreşimi gibi bu; oldukları yerde hafifçe hareket ediyorlar) ki bizim kuvvet alanına en uygun şekli alabilsinler.
- Çözelti Dengelenmesi: Biz bu su ve iyonları eklerken onları mümkün olduğunca üst üste gelmeyecek şekilde yerleştirdik. Bunu yapabilmenin en iyi yolu ise bir ağ dizgesi ya da ızgara(grid) şeklinde dizmektir. O nedenledir ki üstteki resimde bu moleküller sıra sıra dizili gibiler, özellikle su molekülleri proteini bir kılıf gibi sarmıyorlar. Bu fiziki bir durum değil. Çözeltinin protein ile daha sağlıklı bir etkileşime girmesi ve bütün boşlukları doldurması için kısa bir simülasyon yapılır. Bu kısa simülasyonda proteinin henüz hareket etmesini istemiyoruz. O nedenle ekstra bir güç uyguluyoruz. Bu simülasyon sadece cihazları kalibre etmek gibi bir şey o nedenle bu aşamada veri toplanması yapılmıyor.
Veri toplanmasını her molekülün serbest dolaşım hakkına sahip olduğu üretim simülasyonlarından yapıyoruz. Bu simülasyonda proteine uygulanan ekstra gücü kaldırıyoruz. İşte size aşağıda bu simülasyonu gösteriyorum. Pek bir şey olmadığını göreceksiniz, malum ancak 1-2 nanosaniye uzunlukta.Gülün gülün, bende gülüyorum, o bir iki nanosaniyeyi alabilmek için eskiden kaç yıllar geçerdi. Bir deney tüpünde var 1 milyon protein bense gitmiş tek bir tane proteini birkaç nanosaniye kıpraştıracağım diye debeleniyorum.
Öyle bir delilik hali işte bilim. Ama yine de güzel değil mi, insülün ile tanışıp onu sulu bir ortama yerleştirdiniz. İleride simülasyon kısmını detaylandıracağım bir rehberde de koşullar (misal sıcaklık) ile oynayarak neler yapabileciğimizi öğreniriz. Bu alana ilk başladığımda ben böyle film izler gibi izliyordum videoları, keşif yapıyordum. Sonra öğrendim ki meğer grafikler alınıyormuş, her şey matematik her şey istatistikmiş. Şimdi o üstteki resimlere ancak makale yazacağım zaman bakıyorum ben.
Neyse uzun bir rehber oldu ama umarım eğlenmişsinizdir. Bilim ve sanatla kalın! Hoşçakalın. (Aşağıda başka bilgilerde var, ben kaçtım ama siz kaçmayın devam edin.)
%%html --isolated
<iframe src="Simulasyon.html" allowTransparency="true" scrolling="no" width=100% height=550px> </iframe>
Gerekli programlar ve dosyalar¶
Programlama öğrenmek istiyorsanız Python öğrenmek güzel bir adım. İsterseniz Python'ı tek başına indirip kurabilir isterseniz aşağıdaki program üzerinden kurabilirsiniz.
Kendi bilgisayarınızda benim yaptığım gibi bu rehberi çalıştırmak, başka rehberler oluşturmak için Jupyter Notebook kurabilirsiniz. Bu rehberdeki tüm bileşenler Python ve Jupiter Notebook da dahil Anaconda'da mevcut. Tavsiyem Anaconda kurmanız. Komut istemi, kurma etme vs. de yeterli olmama rağmen bütün bu programlar ve gereklilikleri ile tek tek ilgilenmek çok vakit kaybettirici bir iş. O nedenle ben de dahil bir çok kişi Anaconda gibi gibi uygulamalar kullanıyor. Anacondayı kurduktan sonra ek olarak yüklemeniz gereken paketler ise şöyle:
conda install -c bioconda gromacs
conda install -c bioconda nglview
Biraz yorucu olabilir bu işe başlamak, korkutabilir. Bir çok işlemi komut isteminden yapmak daha kolay ama ilk adımı atıp başlaması zor olabilir. Hiç çekinmeyin! Tavsiyem şimdiden elinizi bulaştırıp bu işe komut isteminden girişmeniz.
Benim hazırladığım bu rehberi ve tüm dosyaları github.com/ozyo/MDRehber adresinden indirebilirsiniz. Rehber henüz emekleme aşamadında ama vaktim oldukça güncelleyeceğim. Yorumlarınız veya önerileriniz varsa bana ozgeyoluk_at_proteinart.net adresinden veya EpiSTEM Türkiye üzerinden ulaşabilirsiniz.